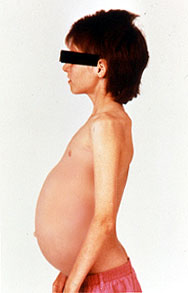
Gaucher's disease is a genetic disease in which a fatty substance (lipid) accumulates in cells and certain organs. Gaucher's disease is the most common of the lysosomal storage diseases. It is caused by a hereditary deficiency of the enzyme glucocerebrosidase (also known as acid β-glucosidase). The enzyme acts on a fatty substance glucocerebroside (also known as glucosylceramide). When the enzyme is defective, the substance accumulates, particularly in cells of the mononuclear cell lineage. Fatty material can collect in the spleen, liver, kidneys, lungs, brain and bone marrow. Symptoms may include enlarged spleen and liver, liver malfunction, skeletal disorders and bone lesions that may be painful, severe neurologic complications, swelling of lymph nodes and (occasionally) adjacent joints, distended abdomen, a brownish tint to the skin, anemia, low blood platelets and yellow fatty deposits on the white of the eye (sclera). Persons affected most seriously may also be more susceptible to infection. The disease is caused by a recessive mutation in a gene located on chromosome 1 and affects both males and females.
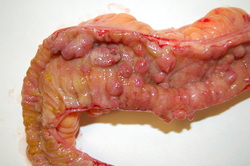
Crohn's disease is a complex, chronic disorder that primarily affects the digestive system,. This condition typically involves abnormal inflammation of the intestinal walls, particularly in the lower part of the small intestine (the ileum) and portions of the large intestine (the colon). Inflammation can occur in any part of the digestive system, however. The inflamed tissues become thick and swollen, and the inner surface of the intestine may develop open sores (ulcers). The disease is caused by a mutation in a gene located on chromosome 2 and affects both males and females.
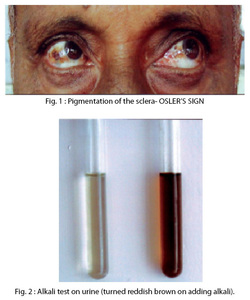
Alkaptonuria is an inherited condition that causes urine to turn black when exposed to air. Ochronosis, a buildup of dark pigment in connective tissues such as cartilage and skin, is also characteristic of the disorder. This blue-black pigmentation usually appears after age 30. People with alkaptonuria typically develop arthritis, particularly in the spine and large joints, beginning in early adulthood. Other features of this condition can include heart problems, kidney stones, and prostate stones. The disease is caused by a mutation in a HGD gene located on chromosome 3.
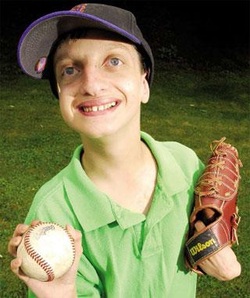
Wolf-Hirschhorn syndrome is a condition that affects many parts of the body. The major features of this disorder include a characteristic facial appearance, delayed growth and development, intellectual disability, and seizures. It is caused by a deletion of genetic material near the end of the short (p) arm of chromosome 4.
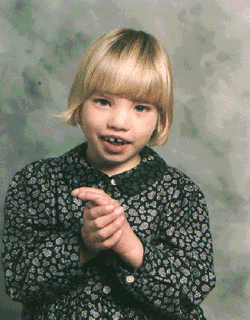
Cri du chat syndrome, also known as chromosome 5p deletion syndrome, 5p- (said minus) syndrome or Lejeune’s syndrome, is a rare genetic disorder due to a missing part (deletion) of chromosome 5. Its name is a French term (cat-cry or call of the cat) referring to the characteristic cat-like cry of affected children
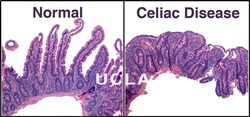
Celiac Disease is a condition in which the immune system is abnormally sensitive to gluten, a protein found in wheat, rye, and barley. Celiac disease is an autoimmune disorder; autoimmune disorders occur when the immune system malfunctions and attacks the body's own tissues and organs. Without a strict, lifelong gluten-free diet, inflammation resulting from immune system overactivity may cause a wide variety of signs and symptoms involving many parts of the body. Celiac
disease is a common disorder. Its prevalence has been estimated at
about 1 in 100 people worldwide. The
risk of developing celiac disease is increased by certain variants of
the HLA-DQA1
and
HLA-DQB1genes found in chromosome 6.
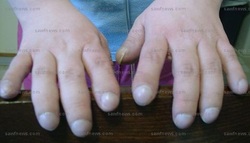
Cystic fibrosis is an inherited disease characterized by the buildup of thick, sticky mucus that can damage many of the body's organs. The disorder's most common signs and symptoms include progressive damage to the respiratory system and chronic digestive system problems. The features of the disorder and their severity varies among affected individuals. Cystic
fibrosis is a common genetic disease within the Caucasian (white)
population in the United States. The disease occurs in 1 in 2,500 to
3,500 Caucasian newborns. Cystic fibrosis is less common in other
ethnic groups, affecting about 1 in 17,000 African Americans and 1 in
31,000 Asian Americans. Mutations
in the CFTR
gene
cause cystic fibrosis in chromosome 7.

Russell-Silver syndrome is a growth disorder characterized by slow growth before and after birth. Babies with this condition have a low birth weight and often fail to grow and gain weight at the expected rate (failure to thrive). Head growth is normal, however, so the head may appear unusually large compared to the rest of the body. Research has focused on genes located in particular regions of chromosome 7 and chromosome 11.
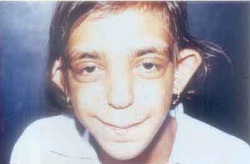
Langer-Giedion syndrome is caused by a deletion or mutation in several genes on the long (q) arm of chromosome 8 at a position described as 8q24.1. This condition causes bone abnormalities, including noncancerous bone tumors known as exostoses, and distinctive facial features. The signs and symptoms of this condition are related to the deletion or mutation in at least two genes from this part of the chromosome. Researchers have determined that the loss of a functional EXT1 gene is responsible for the multiple noncancerous (benign) bone tumors called exostoses seen in people with Langer-Giedion syndrome. Loss of a functional TRPS1 gene may cause the other bone and facial abnormalities. One copy of the EXT1 gene and the TRPS1 gene are always missing or mutated in affected individuals; however, neighboring genes may also be involved. The loss of additional genes from this region of chromosome 8 likely contributes to the varied features of Langer-Giedion syndrome.
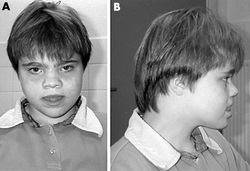
Most people with Kleefstra syndrome, a disorder with signs and symptoms involving many parts of the body, are missing a sequence of about 1 million DNA building blocks (base pairs) on one copy of chromosome 9 in each cell. The deletion occurs near the end of the long (q) arm of the chromosome at a location designated q34.3, a region containing a gene called EHMT1. Some affected individuals have shorter or longer deletions in the same region. The loss of the EHMT1 gene from one copy of chromosome 9 in each cell is believed to be responsible for the characteristic features of Kleefstra syndrome in people with the 9q34.3 deletion. However, the loss of other genes in the same region may lead to additional health problems in some affected individuals.
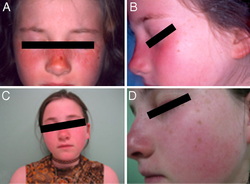
UV-sensitive syndrome is a condition that is characterized by sensitivity to the ultraviolet (UV) rays in sunlight. Even a small amount of sun exposure can cause a sunburn in affected individuals. In addition, these individuals can have freckles, dryness, or changes in coloring (pigmentation) on sun-exposed areas of skin after repeated exposure. Some people with UV-sensitive syndrome have small clusters of enlarged blood vessels just under the skin (telangiectasia), usually on the cheeks and nose. Although UV exposure can cause skin cancers, people with UV-sensitive syndrome do not have an increased risk of developing these forms of cancer compared with the general population. UV-sensitive
syndrome can result from mutations in the ERCC6
gene
(also known as the CSB
gene),
theERCC8
gene
(also known as the CSA
gene),
or the UVSSA
gene found in chromosome 10.
These genes provide instructions for making proteins that are
involved in repairing damaged DNA. DNA can be damaged by UV rays from
the sun and by toxic chemicals, radiation, and unstable molecules
called free radicals. Cells are usually able to fix DNA damage before
it causes problems. If left uncorrected, DNA damage accumulates,
which causes cells to malfunction and can lead to cell death.
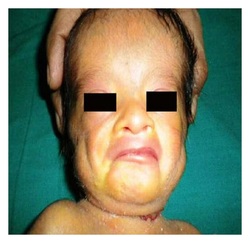
Emanuel syndrome is caused by the presence of extra genetic material from chromosome 11 and chromosome 22 in each cell. In addition to the usual 46 chromosomes, people with Emanuel syndrome have an extra (supernumerary) chromosome consisting of a piece of chromosome 22 attached to a piece of chromosome 11. The extra chromosome is known as a derivative 22 or der(22) chromosome. People
with Emanuel syndrome typically inherit the der(22) chromosome from
an unaffected parent. The parent carries a chromosomal rearrangement
between chromosomes 11 and 22 called a balanced translocation. No
genetic material is gained or lost in a balanced translocation, so
these chromosomal changes usually do not cause any health problems.
As the translocation is passed to the next generation, it can become
unbalanced. Individuals with Emanuel syndrome inherit an unbalanced
translocation between chromosomes 11 and 22 in the form of a der(22)
chromosome. These individuals have two normal copies of chromosome
11, two normal copies of chromosome 22, and extra genetic material
from the der(22) chromosome.
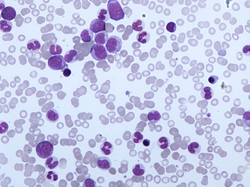
Translocations involving chromosome 12 are involved in a type of blood cell cancer called PDGFRB-associated chronic eosinophilic leukemia. This condition is characterized by an increased number of eosinophils, a type of white blood cell. The most common translocation that causes this condition fuses part of the PDGFRB gene from chromosome 5 with part of the ETV6 gene from chromosome 12, written as t(5;12)(q31-33;p13). Translocations that fuse the PDGFRB gene with other genes can also cause PDGFRB-associated chronic eosinophilic leukemia, but these translocations are relatively uncommon. These translocations are acquired during a person's lifetime and are present only in cancer cells. This type of genetic change, called a somatic mutation, is not inherited.
|
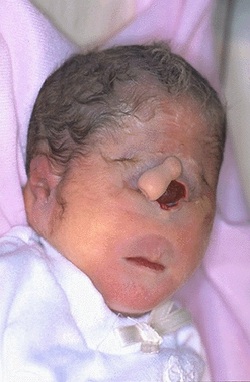
Patau's syndrome is also called Trisomy 13, a chromosomal condition associated with severe intellectual disability and physical abnormalities in many parts of the body. Individuals with trisomy 13 often have heart defects, brain or spinal cord abnormalities, very small or poorly developed eyes (microphthalmia), extra fingers and/or toes, an opening in the lip (a cleft lip) with or without an opening in the roof of the mouth (a cleft palate), and weak muscle tone (hypotonia). Due to the presence of several life-threatening medical problems, many infants with trisomy 13 die within their first days or weeks of life. Only five percent to 10 percent of children with this condition live past their first year.
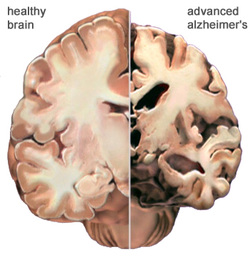
Alzheimer's disease is a degenerative disease of the brain that causes dementia, which is a gradual loss of memory, judgment, and ability to function. This disorder usually appears in people older than age 65, but less common forms of the disease appear earlier in adulthood. Memory loss is the most common sign of Alzheimer disease. Forgetfulness may be subtle at first, but the loss of memory worsens over time until it interferes with most aspects of daily living. Even in familiar settings, a person with Alzheimer disease may get lost or become confused. Routine tasks such as preparing meals, doing laundry, and performing other household chores can be challenging. Additionally, it may become difficult to recognize people and name objects. Affected people increasingly require help with dressing, eating, and personal care. Alzheimer disease currently affects an estimated 2.4 million to 4.5 million Americans. Because the risk of developing Alzheimer disease increases with age and more people are living longer, the number of people with this disease is expected to increase significantly in coming decades. Most cases of early-onset Alzheimer disease are caused by gene mutations that can be passed from parent to child. Researchers have found that this form of the disorder can result from mutations in one of three genes: APP, PSEN1, or PSEN2. When any of these genes is altered, large amounts of a toxic protein fragment called amyloid beta peptide are produced in the brain found in chromosome 14.
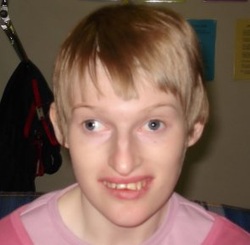
Angelman syndrome results from a loss of gene activity (expression) in a specific part of chromosome 15 in each cell. This region is located on the long (q) arm of the chromosome and is designated 15q11-q13. This region contains a gene called UBE3A that, when mutated or absent, likely causes the characteristic neurologic features of Angelman syndrome. People normally inherit one copy of the UBE3A gene from each parent, and both copies of this gene are turned on (active) in many of the body's tissues. In certain areas of the brain, however, only the copy inherited from a person's mother (the maternal copy) is active. This parent-specific gene activation results from a phenomenon called genomic imprinting. If the maternal copy is lost because of a chromosomal change or a gene mutation, a person will have no working copies of the UBE3A gene in some parts of the brain.
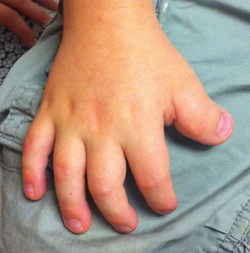
Some cases of severe Rubinstein-Taybi syndrome (also known as chromosome 16p13.3 deletion syndrome) have resulted from a deletion of genetic material from the short (p) arm of chromosome 16. When this deletion is present in all of the body's cells, it can cause serious complications such as a failure to gain weight and grow at the expected rate (failure to thrive) and an increased risk of life-threatening infections. Affected individuals also have many of the typical features of Rubinstein-Taybi syndrome, including intellectual disability, distinctive facial features, and broad thumbs and first toes. Infants born with the severe form of this disorder usually survive only into early childhood. Several genes are missing as a result of the deletion in the short arm of chromosome 16. The deleted region includes the CREBBP gene, which is often mutated or missing in people with the typical features of Rubinstein-Taybi syndrome. Researchers believe that the loss of additional genes in this region probably accounts for the serious complications associated with severe Rubinstein-Taybi syndrome.

Deletion of a small amount of genetic material (a microdeletion) on chromosome 17 can cause Koolen-de Vries syndrome. This disorder is characterized by developmental delay, intellectual disability, a cheerful and sociable disposition, and a variety of physical abnormalities. Most people with Koolen-de Vries syndrome are missing a sequence of about 500,000 base pairs, also written as 500 kilobases (kb), at position q21.31 on chromosome 17. The exact size of the deletion varies among affected individuals, but it contains at least six genes including KANSL1. This deletion affects one of the two copies of chromosome 17 in each cell.
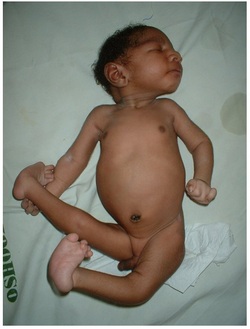
Trisomy 18, also called Edwards syndrome, is a chromosomal condition associated with abnormalities in many parts of the body. Individuals with trisomy 18 often have slow growth before birth (intrauterine growth retardation) and a low birth weight. Affected individuals may have heart defects and abnormalities of other organs that develop before birth. Other features of trisomy 18 include a small, abnormally shaped head; a small jaw and mouth; and clenched fists with overlapping fingers. Due to the presence of several life-threatening medical problems, many individuals with trisomy 18 die before birth or within their first month. Five to 10 percent of children with this condition live past their first year, and these children often have severe intellectual disability.
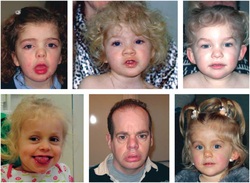
Coffin-Siris syndrome is a condition that affects several body systems. Although there are many variable signs and symptoms, hallmarks of this condition include developmental disability, abnormalities of the fifth (pinky) fingers or toes, and characteristic facial features. Most affected individuals have mild to severe intellectual disability or delayed development of speech and motor skills such as sitting and walking. Another feature of Coffin-Siris syndrome is underdevelopment (hypoplasia) of the tips of the fingers or toes, or hypoplasia or absence of the nails. These abnormalities are most common on the fifth fingers or toes. In addition, most affected individuals have facial features described as coarse. These typically include a wide nose with a flat nasal bridge, a wide mouth with thick lips, and thick eyebrows and eyelashes. Affected individuals can have excess hair on other parts of the face and body (hirsutism), but scalp hair is often sparse. There is a range of facial features seen in people with Coffin-Siris syndrome, and not all affected individuals have the typical features. In addition, people with this condition may have an abnormally small head (microcephaly). Coffin-Siris syndrome is a rare condition that is diagnosed in females more frequently than in males. Approximately 80 cases have been reported in the medical literature. Coffin-Siris syndrome is caused by mutations in the ARID1A, ARID1B, SMARCA4, SMARCB1, or SMARCE1 gene found in chromosome 19.
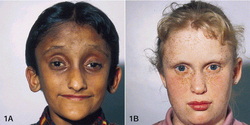
Approximately 7 percent of individuals with Alagille syndrome have small deletions of genetic material on chromosome 20, in a region known as 20p12. This region includes the JAG1 gene, which is involved in signaling between neighboring cells during embryonic development. This signaling influences how the cells are used to build body structures in the developing embryo. Loss of the JAG1 gene probably disrupts the signaling pathway. As a result, errors may occur during development, especially affecting the heart, bile ducts in the liver, the spinal column, and certain facial features. The
estimated prevalence of Alagille syndrome is 1 in 70,000 newborns.
This figure is based on diagnoses of liver disease in infants, and
may be an underestimation because some people with Alagille syndrome
do not develop liver disease during infancy.
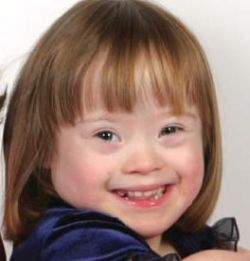
Down syndrome is a chromosomal condition that is associated with intellectual disability, a characteristic facial appearance, and weak muscle tone (hypotonia) in infancy. This condition is most often caused by trisomy 21. Trisomy 21 means that each cell in the body has three copies of chromosome 21 instead of the usual two copies. Less commonly, Down syndrome occurs when part of chromosome 21 becomes attached (translocated) to another chromosome during the formation of reproductive cells (eggs and sperm) or very early in fetal development. Affected people have two copies of chromosome 21 plus extra material from chromosome 21 attached to another chromosome, resulting in three copies of genetic material from chromosome 21. Affected individuals with this genetic change are said to have translocation Down syndrome.
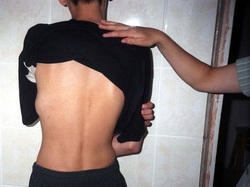
Translocations involving chromosome 22 are also involved in a type of cancerous tumor known as Ewing sarcoma. These tumors develop in the bones or soft tissues, such as cartilage and nerves. The most common translocation, t(11;22), fuses part of the EWSR1 gene from chromosome 22 with part of the FLI1 gene from chromosome 11, creating the EWSR1/FLI1 fusion gene. Translocations that fuse the EWSR1 gene with other genes that are related to the FLI1 gene can also cause Ewing sarcomas, although these alternative translocations are relatively uncommon. The mutations that cause these cancers are acquired during a person's lifetime and are present only in tumor cells. This type of genetic change, called a somatic mutation, is not inherited.
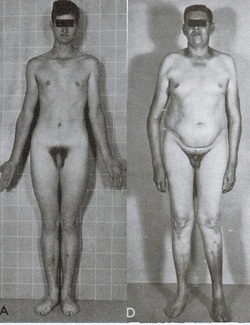
Klinefelter syndrome is caused by the presence of one or more extra copies of the X chromosome in a male's cells. Extra genetic material from the X chromosome interferes with male sexual development, preventing the testes from functioning normally and reducing the levels of testosterone (a hormone that directs male sexual development). A shortage of testosterone can lead to delayed or incomplete puberty, genital abnormalities, breast enlargement (gynecomastia), reduced facial and body hair, and an inability to have biological children (infertility). Children with Klinefelter syndrome may also have learning disabilities, delayed speech and language development, and a shy and unassuming personality. Typically, people with Klinefelter syndrome have one extra copy of the X chromosome in each cell, for a total of two X chromosomes and one Y chromosome (47,XXY). Less commonly, affected individuals may have two or three extra X chromosomes (48,XXXY or 49,XXXXY). As the number of extra sex chromosomes increases, so does the risk of learning problems, intellectual disability, birth defects, and other health issues.
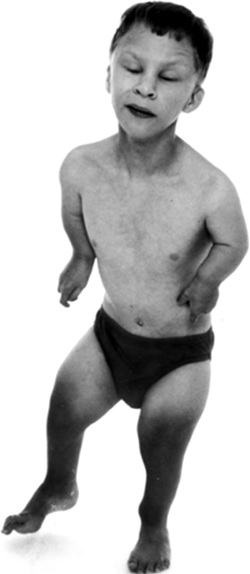
Langer mesomelic dysplasia is a disorder of bone growth. Affected individuals typically have extreme shortening of the long bones in the arms and legs (mesomelia). As a result of the shortened leg bones, people with Langer mesomelic dysplasia have very short stature. A bone in the forearm called the ulna and a bone in the lower leg called the fibula are often underdeveloped or absent, while other bones in the forearm (the radius) and lower leg (the tibia) are unusually short, thick, and curved. Some people with Langer mesomelic dysplasia also have an abnormality of the wrist and forearm bones called Madelung deformity, which may cause pain and limit wrist movement. Additionally, some affected individuals have mild underdevelopment of the lower jaw bone (mandible). Langer mesomelic dysplasia results from changes involving the SHOX gene found in Y chromosome.
|
